
Wie kann ich mich auf etwas vorbereiten, das ich noch nicht weiß?
Wissenschaftler der Technischen Universität München (TUM) und des Fritz-Haber-Instituts Berlin haben sich dieser fast philosophischen Frage im Kontext des maschinellen Lernens gewidmet.
Lernen bedeutet nichts anderes, als neue Entscheidungen auf der Grundlage früherer Erfahrungen zu treffen. Um auf diese Weise mit einer neuen Situation umgehen zu können, muss man sich bereits zuvor mit annähernd ähnlichen Situationen auseinandergesetzt haben. Beim maschinellen Lernen bedeutet dies entsprechend, dass ein Lernalgorithmus in etwa ähnlichen Daten ausgesetzt gewesen sein muss.
Aber was können wir tun, wenn die Möglichkeiten nahezu unendlich sind und es schlicht unmöglich ist, Daten zu generieren, die alle Situationen abdecken? Dieses Problem wird zu einer großen Herausforderung, wenn man es mit einer endlosen Anzahl möglicher Kandidatenmoleküle zu tun hat.
Organische Halbleiter ermöglichen wichtige Zukunftstechnologien wie tragbare Solarzellen oder rollbare Displays. Für solche Anwendungen müssen verbesserte organische Moleküle entdeckt werden, aus denen diese Materialien bestehen. Bei Aufgaben dieser Art kommen zunehmend Methoden des maschinellen Lernens zum Einsatz, während das Training auf Daten aus Computersimulationen oder Experimenten basiert.
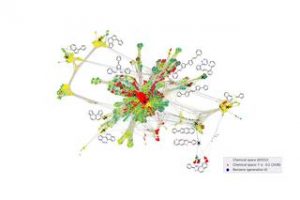
Die Zahl potenziell möglicher kleiner organischer Moleküle wird jedoch auf etwa 1033 geschätzt. Diese überwältigende Zahl an Möglichkeiten macht es praktisch unmöglich, genügend Daten zu generieren, um eine so große Materialvielfalt abzubilden. Zudem sind viele dieser Moleküle nicht einmal für organische Halbleiter geeignet. Man sucht quasi nach der sprichwörtlichen Nadel im Heuhaufen.
Das Team um Prof. Karsten Reuter, Leiter der Theorieabteilung am Fritz-Haber-Institut, und Dr. Harald Oberhofer, Heisenberg-Stipendiat am Lehrstuhl für Theoretische Chemie, ging dieses Problem durch sogenanntes aktives Lernen an. Anstatt aus vorhandenen Daten zu lernen, entscheidet der Machine-Learning-Algorithmus iterativ selbst, welche Daten er tatsächlich benötigt, um über das Problem zu lernen.
Die Wissenschaftler führen zunächst Simulationen an einigen kleineren Molekülen durch und erhalten Daten zur elektrischen Leitfähigkeit der Moleküle – ein Maß für deren Nützlichkeit bei der Untersuchung möglicher Solarzellenmaterialien.
Basierend auf diesen Daten entscheidet der Algorithmus, ob bereits kleine Modifikationen an diesen Molekülen zu nützlichen Eigenschaften führen könnten oder ob dies aufgrund fehlender ähnlicher Daten unsicher ist. In beiden Fällen fordert es automatisch neue Simulationen an, verbessert sich durch die neu generierten Daten, berücksichtigt neue Moleküle und wiederholt diesen Vorgang.
In ihrer Arbeit zeigen die Wissenschaftler, dass dieser Ansatz deutlich effizienter ist als alternative Suchalgorithmen und wie auf diese Weise neue, vielversprechende Moleküle identifiziert werden können, während der Algorithmus seine Erkundung des riesigen Molekülraums fortsetzt. Jede Woche werden neue Moleküle vorgeschlagen, die die nächste Generation von Solarzellen einläuten könnten, und der Algorithmus wird immer besser.